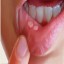
All About Microtia and Aural Atresia

As the father of a wonderful three-year-old son (Emmanuel) who was born with Microtia and Aural Atresia, I will be the first person to admit that neither my wife Theresa, nor myself, knew anything about the little known syndrome that is believed to occur once in every 6,000 to 12,000 births.
However, it wouldn’t be long before we both began to research Microtia and speak with several medical professionals in an effort to learn everything about Microtia that we could. Having dealt with all of the emotions three years ago, not to mention the initial shock, my wife and I realize that each passing day brings Emmanuel – and us – one day closer to jubilation. At the same time, we both thought it would a wonderful idea to share some of our knowledge and some facts about the little known condition called Microtia.
What is Microtia?
The term microtia indicates a small, abnormally shaped or absent external ear. It can occur on one side only (called “unilateral”) or on both sides (called “bilateral”). The unilateral form is much more common, occurring in approximately 90% of patients.
What is Aural Atresia?
The term aural atresia refers to the absence of the ear canal. Patients who have microtia usually, but not always, also have aural atresia. Patients who have aural atresia have no hearing on that side but usually have completely normal hearing in the normal ear. Obviously, patients who have atresia in both ears will be sufficiently hard of hearing to require a hearing aid. (See below).
Patients who lack the ear canal also have structural abnormalities of the middle ear with absence of the eardrum and incomplete formation of the small middle ear bones, which allow conduction of hearing through the middle ear. Microtia and aural atresia tend to occur together because the outer ear and the middle ear evolve from a common embryologic origin.
Is Microtia an Isolated Condition?
In most patients, microtia and aural atresia occur as an isolated condition. In some patients, however, the ear deformity occurs in conjunction with other facial abnormalities. The most common condition in which microtia accompanies other anomalies is called “hemifacial microsomia”. This condition has several other names, which makes it difficult to read about for the layperson. For example, hemifacial microsomia is also known as the “1st and 2nd branchial arch syndrome” and the “oto-mandibular syndrome” as well as by other names. This condition can be quite variable but involves underdevelopment of all the structures on one side of the face, including the ear, the bones of the face, and the fullness of the cheek tissue and the function of the facial nerve. Like isolated microtia, hemifacial microsomia can also occur on both sides of the face, which occurs in a minority of patients.
Another syndrome where microtia may be present is Treacher Collins Syndrome. This condition is always bilateral and tends to be an inherited syndrome and involves underdevelopment of the lower eyelids, the cheekbones, and the lower jaw. Patients who have severe manifestations of the syndrome frequently have upper airway obstruction and require a tracheostomy and/or surgical distraction of the lower jaw at a very young age.
Because of the association with these other conditions, patients with microtia may require access to other specialists and may require a genetics evaluation so that parents can be informed of any increase risk of these conditions in future children.
What Should We Do if We Have a Child With Microtia?
In most patients, the only issue, which should be addressed early in childhood, is an evaluation of the hearing. As described above, patients, who have involvement of one side of the face only, almost always have normal hearing in the other ear and will consequently have essentially normal hearing. It is important; however, to ensure that there is sufficient hearing early in childhood because children will not develop normal speech unless they have normal hearing. A hearing test (called an audiogram) will be recommended shortly after birth. There are two types of hearing tests, BAER testing (brain stem auditory evoked response testing) and Behavioral Testing. BAER testing is performed before the child is old enough to cooperative with behavioral testing. Reliable behavioral testing is the procedure of choice when the child is mature enough to cooperate.
To reiterate, patients with a unilateral deformity usually have normal hearing in the opposite ear and do not require a hearing aid. In the minority of patients, however, the condition affects both ears and these patients will require a hearing aid called a “bone conduction” hearing aid.
What if Sufficient Hearing Does Not Exist?
The bone conduction hearing aids will be recommended within the first few months of life. The evaluation and surgical reconstruction of the hearing will not be recommended for several years. Unfortunately, patients with Treacher Collins Syndrome usually have sufficient anatomic abnormalities of the middle ear that the hearing cannot be surgical reconstructed and these patients are dependent on a hearing aid for life. The Food and Drug Administration in the United States have recently approved a relatively new type of hearing aid called the “bone anchored hearing aid” (BAHA) which is a small box attached to a small screw imbedded in the skull. This can be placed within the hair so it is not conspicuous and tends to provide better amplification of hearing than other hearing aids.
How Often Should Hearing and Language Be Monitored?
Regular monitoring of hearing and language is important and should be performed at least as often as outlined in the attached protocol. If a child develops frequent middle ear infections, “otitis media”, in the normal ear, the need for monitoring of hearing and language will be more frequent. Since patients who have microtia/aural atresia on one side are completely dependent on the other ear for hearing, it is important to make sure that any ear infections in the normal ear are treated completely to maximize the hearing in the better ear. Infections can result in accumulation of fluid in the middle ear, which can significantly reduce hearing. This condition is treatable and therefore regular follow up by a pediatric otolaryngologist is essential.
Do Patients with Microtia/Aural Atresia Require Any Other Tests?
A high resolution, 3-D CT scan of the temporal bones is recommended to rule out a benign tumor of the middle ear known as a cholesteatoma which is more common in atresia patients. In addition, the CT scan will indicate if the middle ear structures can be surgically reconstructed. In other words, all patients with microtia are candidates for surgical reconstruction of the outer ear, but not all patients with aural atresia are candidates for reconstruction of the ear canal and middle ear.
Is Middle Ear Surgery Recommended in the Unilateral Cases?
Patients with aural atresia on one side will have essentially normal hearing based on the opposite ear. The patients may experience difficulty localizing sounds because they lack the stereo effect of having two ears. Because the patients with the unilateral condition have essentially normal hearing, most of these patients do not have reconstruction of the middle ear on the affected side.
On the other hand, the surgical reconstruction of the hearing has improved dramatically over recent years and some families opt to have the middle ear reconstruction on the affected side, even in the unilateral cases. This is an individual patient/family decision, which balances the benefit of having “stereo” hearing vs. an outer ear reconstruction, which will not be quite as pleasing. The surgery to reconstruct the middle ear does affect the aesthetics of the outer ear reconstruction.
At What Age Is the External Ear Reconstruction Initiated?
This is an important question, which is undergoing some evolution. Up until recently, surgical reconstruction of the outer ear was recommended beginning at the age of six years. At this age the patients were thought to have sufficient cartilage in the rib cage to allow reconstruction of the ear. As surgical techniques have improved, however, it is clear that a better quality, more detailed, ear reconstruction is possible when the surgery is delayed to after the age of ten years.
When Is Surgery Performed To Reconstruct the Ear Canal and Middle Ear?
If surgery is recommended for reconstruction of the middle ear, it only takes place after the outer ear has been reconstructed. It is important that the outer ear and middle ear reconstructions be coordinated. If the middle ear procedure is performed first, it may eliminate the chances for reconstruction of the outer ear.
How Many Stages Are Required To Reconstruct an Ear?
Depending on the technique used and on the degree of abnormality, the reconstruction can be performed in two, three, or four stages. There is a movement toward reducing the number of stages in as many patients as possible to a total of two stages. Generally though, it takes three and sometime four, procedures.
What Other Support Is Available?
It is often difficult for parents to deal with their own responses to their child’s condition as well as the responses of other people. It may be helpful to speak to other families who have children with similar conditions. I know the entire Williams family hopes this information can provide some help for families that may be looking for assistance with Microtia-related issues.